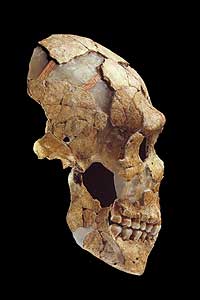
Das Genom des Neandertalers wird entschlüsselt
Seit der ersten Entdeckung eines Neandertalerfossils vor 150 Jahren versuchen Paläontologen und Anthropologen aufzudecken, welche Rolle dieser Frühmensch, der in Europa und weiten Teilen Asiens lebte, in der Evolution des modernen Menschen gespielt hat. Ein Forscherteam um Prof. Svante Pääbo vom Max-Planck-Institut für evolutionäre Anthropologie in Leipzig will nun in Zusammenarbeit mit der us-amerikanischen Firma 454 Life Sciences das gesamte ca. 3 Millionen Basenpaare umfassende Genom des Neandertalers beschreiben. Als ersten Test haben die beiden Projektpartner bereits etwa eine Million Basenpaare der Zellkern-DNA eines 38.000 Jahre alten Neandertalerfossils entschlüsselt. Die Analyse von prähistorischer DNA (aDNA) ist ungleich komplizierter als die Arbeit mit Material aus lebenden Organismen und in jahrzehntelanger Forschung mussten die möglichen Fehlerquellen in den Untersuchungsmethoden identifiziert werden.
Die DNA-Analyse
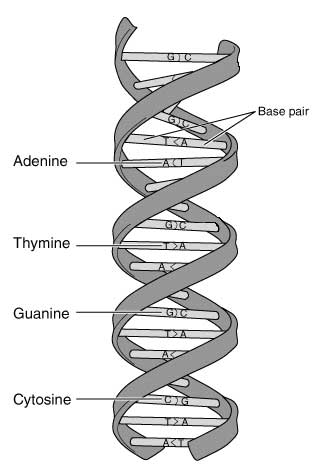
Um zu verstehen, wo die Fallstricke bei der Arbeit speziell mit aDNA versteckt liegen, muss man sich, eigene leidvolle Erfahrung des Autors, mit den Techniken und Methoden der DNA-Analyse auseinandersetzen. Im Grunde genommen hat die DNA (desoxyribonucleic acid) die Struktur einer schraubenförmig gewundenen Strickleiter, die aus zwei gleichen, aber gegenläufig angeordneten DNA-Einzelsträngen besteht, die jeweils ein sogenanntes 5'-Ende mit einer Phosphat-Gruppe und ein 3'-Ende mit einer OH-Gruppe besitzen. Entlang jedes dieser „Leiterholme“ sitzen in mannigfaltiger Kombination die vier Buchstaben des Erbgutes. Diese Buchstaben werden durch ein Nukleotid repräsentiert, das jeweils eine der vier Basen Adenin (A), Thymin (T), Cytosin (C) und Guanin (G) enthält. Jedes dieser Nukleotide verbindet sich zusammen mit dem Gegenstück des gegenüberliegenden Einzelstranges zu einem Basenpaar und somit zu den „Sprossen“ der DNA-Strickleiter. Dabei können sich die Basen nur komplementär paaren: nur Adenin mit Thymin, sowie Cytosin mit Guanin. Diese Tatsache verhindert, dass es bei den unzähligen Duplizierungen der DNA im Laufe des Lebens eines Organismus zu folgenschweren Schreibfehlern kommt.
Im Prinzip könnte man das Auslesen der Nukleotidreihenfolge, das Sequenzieren, mit einem Rastertunnelmikroskop praktizieren. Das würde allerdings Unmengen an Zeit und somit auch Geld in Anspruch nehmen, deshalb basieren die Analysemethoden in vielen Schritten auf chemischen Verfahren.
Der allererste Schritt ist die Extraktion des DNA-Moleküls aus einer Zelle. DNA aus lebenden Organismen muss danach aus verfahrenstechnischen Gründen in viele kleine Stücke getrennt werden. Bei aDNA entfällt dieser Schritt, da sie eh in viele kleine Fragmente zerfallen ist, dazu später mehr.
Die wundersame Vermehrung – die Amplifikation
Weil die Empfindlichkeit der Geräte für die DNA-Sequenzierung zu gering ist, um die Basenpaare eines einzigen DNA-Fragmentes auszulesen, muss es vorher in sehr hoher Stückzahl kopiert werden, die Amplifikation. Dabei werden mit Hilfe der sogenannten Polymerase-Kettenreaktion (abgekürzt PCR, von polymerase chain reaction) meist mehrere Millionen Kopien der jeweiligen Fragmente erzeugt. Für die PCR wird das DNA-Fragment zuerst durch Erhitzen in seine beiden Einzelstränge aufgetrennt. Nun tritt das Enzym Polymerase in Aktion. Es wandert an dem Einzelstrang entlang und führt dazu, dass sich an jeder Base, besser jedem Nukleotid, wieder der Komplementär anlagert und somit ein neuer Doppelstrang entsteht. Das Gleiche erfolgt mit dem anderen Einzelstrang. Ergebnis, ein DNA-Fragment wurde verdoppelt. Die dafür benötigten Basen A, T, C und G werden dem Prozess vorher zugegeben. Dieser Zyklus wird nun mehrere Male durchgeführt und die DNA-Fragmente dabei exponentiell vermehrt. Die PCR kann allerdings nicht einfach am Einzelstrang ansetzen, sondern sie benötigt einen Startpunkt, an dem das zu kopierende DNA-Fragment bereits als Doppelstrang vorliegt. Im Falle des Neandertalers wird jedes aDNA-Einzelstrangfragment einfach mit einer sehr kurzen synthetisch hergestellten Nukleotidsequenz (sehr kurzer DNA-Einzelstrang) verlängert, für die das passende komplementäre Gegenstück, der Primer, vorhanden ist. Am 3'-Ende dieses Gegenstückes kann nun die Polymerase ansetzen und den Doppelstrang komplett neu ausbilden.
Buchstabe für Buchstabe gelesen - die Sequenzierung

Mittlerweile gibt es mehrere unterschiedliche Methoden, wie die einzelnen Sequenzen der DNA, also ihre Buchstaben, analysiert und gelesen werden. Der Weg dahin läuft allerdings bei allen Verfahren über eine erneute Polymerase, diesmal jedoch nicht als Kettenreaktion mit dem Ziel der Vermehrung, sondern kontrolliert und Stück für Stück. Zuerst werden die in der Amplifikation vermehrten DNA-Fragmente erneut in Einzelstränge zerlegt und die Polymerase in Gang gesetzt. Das Verfahren, mit dem das Neandertalergenom entziffert wird, benutzt für die Sequenzierung Bioluminiszenz.
Dem Polymeraseprozess werden Proteine beigegeben, die immer dann Licht produzieren, wenn eine Base an den Gegenstrang der DNA angesetzt wird. Die Lichtproteine werden dabei durch Pyrophosphat angeregt, das während der Verbindung der Basen entsteht. Und damit dieses Lichtsignal messbar wird, braucht man diese Unmengen identischer DNA-Fragmente, die genügend Proteine gleichzeitig zum Aufleuchten bringen. Um nun die Reihenfolge der Basen Ademin, Thymin, Cytosin und Guanin herausfinden zu können, wird der Polymerase jeweils nur eine dieser Basen zur Verfügung gestellt. Leuchtet nichts, hat sich die Base nicht angelagert. Leuchtet es, kann man je nach Lichtintensität feststellen, wie viele Nukleotide der einen Base gebildet wurden. Das Ganze läuft solange, bis alle Basen des DNA-Strangs ihre Partner bekommen haben. Die Buchstaben des ursprünglichen DNA-Fragmentes sind entschlüsselt und lesen sich zum Beispiel so: ...AGGATTAGGCCTG...
Von Bakterien, Pilzen und Wissenschaftlern
Die aDNA von Fossilien zu analysieren ist eine technische Herausforderung. Wenn ein Organismus stirbt, werden seine Zellen von Bakterien und Pilzen überrannt. Ein Großteil seiner DNA wird zerstört und der geringe Teil, der noch vorhanden ist, zerfällt während der langen Dauer der Fossilierung in kleine Stücke und wird zu allem Übel noch chemisch verändert. Allgemein gehen die Wissenschaftler davon aus, dass bei einer Extraktion aus fossilen Knochen nur sehr wenige Fragmente der ursprünglichen DNA vorhanden sind, oftmals nur ein einziges. Generell sind die aDNA-Fragmente in einer Länge von 200 Basenpaaren erhalten, im Vergleich dazu, die DNA aus einer Zelle lebender Menschen hat 3 Milliarden. Wenn Wissenschaftler also DNA-Bruchstücke aus einem fossilen Knochen extrahieren konnten, sind diese zum einen winzig und zum anderen stammt der Großteil von Kontaminanten wie Bakterien, Pilzen und nicht selten den Wissenschaftlern, die zuvor mit den Knochen zu tun hatten. Dies sind alles keine perfekten Umstände zur Erforschung prähistorischer DNA.
Deshalb konzentrierte man sich bei der Erforschung alter DNA bisher meist auf die mitochondriale DNA (mtDNA), einem kleinen DNA-Ring, der in den Mitochondrien der Zellen zu finden ist. Diese liegt im Vergleich zur DNA des Zellkerns in wesentlich größerer Kopienzahl vor (etwa 1000 mitochondriale Kopien pro Zelle, aber nur 2 im Zellkern), wodurch die oben genannten Erhaltungsprobleme etwas gelindert werden. Allerdings repräsentiert die mtDNA nur etwa 0,001% des gesamten Genoms eines Säugetiers und wird ausschließlich über die mütterliche Linie vererbt. Daher vermittelt sie nur begrenzte Einsichten, wie sehr sich alte von heute lebenden Organismen unterscheiden. Aus diesem Grund soll nun in dem zweijährigen Projekt, dass von der Max-Planck-Gesellschaft finanziert wird, das gesamte Genom des Neandertalers aus der Zellkern-DNA entschlüsselt werden.
Und wieder nichts – die Probleme mit prähistorischer DNA
Ein generelles Problem war bisher die Menge des benötigten Materials. Bis zur Einführung des neuen Untersuchungsverfahrens von 454 Life Sciences, wären für die Entschlüsselung des gesamten Genoms mehrere Kilogramm Neandertalerknochen notwendig gewesen, um die erforderlichen Millionen von PCR-Reaktionen durchführen zu können. Nun kann man Museumsdirektoren noch davon überzeugen, kleinste Proben aus „ihren“ Neandertalerknochen entnehmen zu dürfen, aber man ringt ihnen mit Sicherheit kein Einverständnis für die Pulverisierung des gesamten Skelettmaterials ab. Da mit der neuen Methode auch die winzigen aDNA-Fragmente aus kleinen Mengen Knochenmaterial in ausreichender Zahl amplifiziert und sequenziert werden können, steht dem Unterfangen des internationalen Forschungsteams zumindest kein Museumsdirektor skeptisch gegenüber.
Die sichtbare Erhaltung eines Organismus sagt wenig über den Zustand der enthaltenen aDNA aus. Aus Moorleichen ist beispielsweise aufgrund des sauren Liegemilieus selten verwertbare DNA zu extrahieren. Ob eine Knochenprobe überhaupt verwertbare DNA-Fragmente enthält, wird deshalb schon im Vorfeld durch biochemische Tests überprüft. Häufig untersucht man dazu die Aminosäuren. Sind diese bis zu einem gewissen Grad erhalten, kann man mit großer Sicherheit davon ausgehen, auch alte DNA zu finden. Weitermachen lohnt sich also bei so einer Probe.

Ebenso haben sich durch die jahrzehntelange Arbeit mit aDNA Kriterien herausgebildet, die schon während der Amplifikation und der Sequenzierung auf die Existenz von aDNA, bzw. auf deren mögliche Verschmutzungen durch Fremd-DNA hinweisen können. Von alter DNA sollten eigentlich nur kurze Stücke amplifiziert werden, da diese DNA hochgradig zerfallen ist. Längere DNA-Stücke sind eher ein Hinweis auf jüngere DNA. In eine ähnliche Richtung zielt die Überlegung, die Zahl der notwendigen PCR-Zyklen als Indikator zu verwenden. Die Ausgangsmenge an aDNA ist auf Grund ihres weit fortgeschrittenen Zerfalls immer sehr gering und benötigt somit deutlich mehr Zyklen, bis genügend Untersuchungsmaterial entstanden ist. Somit liegt bei Proben, die häufiger amplifiziert werden müssen, die Vermutung nahe, dass es sich hierbei um aDNA handelt.
Doch selbst wenn diese Kriterien erfüllt sind, halten die Zerfallsprozesse in den Fossilien weitere Fallgruben für die Genetiker bereit. Zum Beispiel kann sich mtDNA in die DNA des Zellkerns integrieren. Dies führt unweigerlich zu Fehleinschätzungen, besonders bei der Analyse von Verwandtschaftsgraden. In eine ähnliche Richtung gehen falsche Kopiervorgänge bei den PCR-Prozessen. Grund dafür sind, sie erraten es, die im Laufe der Jahrtausende auftretenden chemischen Veränderungen der Nukleotiden an einzelnen Stellen in der DNA. Bei der Polymerase können dadurch falsche Basen eingebaut und somit Pseudo-Mutationen erzeugt werden. An solchen Stellen, gerne „Hot Spots“ genannt, werden hauptsächlich T statt C und A statt G eingebaut. Nur durch viele Vergleiche kann man hier erkennen, welche ausgelesene Sequenz nun die richtige ist. Zur Kontrolle müssen und sollen Proben immer auch in anderen Laboratorien untersucht werden und die Ergebnisse natürlich identisch sein.
Vergleichen, Vergleichen und nochmals Vergleichen
Und nur durch viele Vergleiche rückt man den Bakterien, Pilzen und Wissenschaftlern zu Leibe, die im schlimmsten Falle alle mit ihren genetischen Spuren in den Analyseergebnissen auftauchen. Denn selbst unter den besten Umständen liefert ein sehr großer Teil der sequenzierten Proben DNA, die nicht vom Neandertaler stammt. Das heißt, es müssen eine Unmenge an einzelnen Sequenzierungen vorgenommen werden, dazu kommen noch zahlreiche Kontrolldurchgänge, um zu überprüfen, ob die gleiche Probe immer zu gleichen Ergebnissen führt. Und hier ist ein weiterer großer Vorteil der neuen Untersuchungsmethode. Sie ist schnell. Ein Gerät liefert in etwa 12 Stunden bis zu 200.000 verwertbare DNA-Sequenzen.
Doch wie kommt das Forschungsteam um Svante Pääbo jetzt, trotz all der geschilderten Probleme, zu der sehnlichst erwünschten Neandertaler-DNA? Das ist einfach erklärt: durch Vergleichen, Vergleichen und nochmals Vergleichen. Alle ausgelesenen und aufgeschlüsselten DNA-Sequenzen werden mit anderen schon bekannten Sequenzen verglichen, um die Kontaminationen zum Beispiel von Bakterien etc. herauszufiltern. Abgesehen davon, dass Bakterien und Pilze eh sehr signifikante DNA-Sequenzen haben. Am schwierigsten gestaltet sich natürlich die Unterscheidung der DNA des Neandertalers und des modernen Menschen. Um dieses Vorgehen zu erklären, müssen wir kurz einen Blick auf die gegenseitigen Verwandtschaftsverhältnisse der folgenden drei Protagonisten werfen. Die Entwicklung der Schimpansen, sowie der Gattung Mensch trennte sich vor knapp 6 Millionen Jahren. Die Schimpansen sind mit 99% übereinstimmender DNA die nächsten lebenden Verwandten des heutigen Menschen. Im gleichen Bereich muss die Überstimmung zwischen Schimpanse und Neandertaler liegen, unserem engsten, allerdings ausgestorbenen Verwandten. Die Linien Neandertaler und moderner Mensch haben sich vor knapp 500.000 Jahren getrennt. Die Unterschiede zwischen dem modernen Mensch und dem Neandertaler liegen somit in dem restlichen einen Prozent. Unsere DNA ist bekannt und die des Schimpansen ist ebenfalls komplett entziffert. Und so werden „einfach“ die sequenzierten Proben mit der DNA des Schimpansen und der des modernen Menschen verglichen. Sequenzen, die weder dem Menschen noch dem Schimpansen gleichen, sind mit allerhöchster Wahrscheinlichkeit vom Neandertaler. Entstanden seit der Zeit seiner Trennung von der Linie des modernen Menschen.
Und dann wird gepuzzelt
In den nächsten zwei Jahren möchten die Wissenschaftler Proben aus mehreren gut erhaltenen Neandertalern entnehmen, um etwa 60 Milliarden Basen aus Millionen von DNA-Fragmenten zu bestimmen. Da sich viele dieser bestimmten Fragmente in größeren Bereichen überlappen, kann man aus ihnen einen Entwurf der 3 Milliarden Basen rekonstruieren, aus denen das Genom des Neandertalers besteht.
Oberste Priorität bei der Erfoschung der aDNA ist im Übrigen die Reinheit des Labors und der Geräte. Zur Kontrolle werden immer wieder sogenannte negative PCR-Zyklen durchgeführt. Dabei wird statt DNA Wasser verwendet. Kommt es zu einer Amplifikation, müssen irgendwo im Laufe des Untersuchungsprozesses fremde DNA-Spuren zu Verschmutzungen führen und von „falscher“ DNA hat man ja nun schon mehr als genug.